

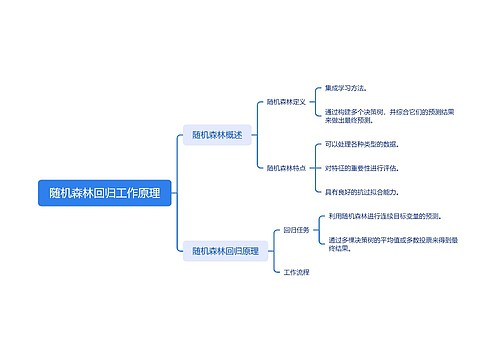
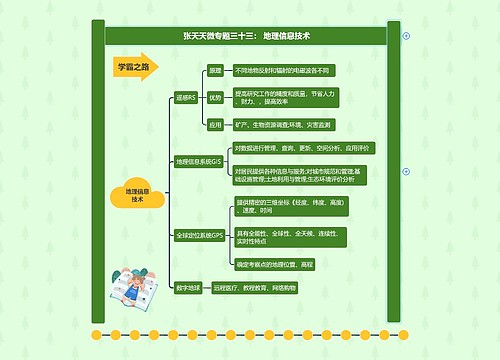
临床药理学的任务思维导图

1 新药的研究与评价 新药的研究过程一般要经过三个阶段,即实验药理、临床前药理和临床药理。在许多国家,新药上市都必须呈报临床前药理、毒理和临床药理研究资料。我国从85年7月1日起,按卫生部《新药审批崐办法》规定,各类新药的呈报资料中必须有临床药理研究结果。
树图思维导图提供 临床药理学的任务 在线思维导图免费制作,点击“编辑”按钮,可对 临床药理学的任务 进行在线思维导图编辑,本思维导图属于思维导图模板主题,文件编号是:e01c666ebb578c7029a2414b2bd0e339
思维导图大纲
临床药理学的任务思维导图模板大纲
1 新药的研究与评价
新药的研究过程一般要经过三个阶段,即实验药理、临床前药理和临床药理。在许多国家,新药上市都必须呈报临床前药理、毒理和临床药理研究资料。我国从85年7月1日起,按卫生部《新药审批崐办法》规定,各类新药的呈报资料中必须有临床药理研究结果。新药的临床药理研究的主要内容为新药临床试验,分四期进行。Ⅰ期临床试验以健康志愿者为受试对象,一般为20~30人,研究新药人体耐受性与药代动力学,为Ⅱ期临床试验提供安全有效的合理试验方案。Ⅱ期临床试验以病人为试验对象,一般10~100例,进行新药与对照药的随机对照临床试验,详细考察新药的疗效、适应症、不良反崐应,对其安全有效性作出确切评价。本期结束后,即可将临床试验结果及临床前药理研究结果汇总,向药政主管部门办理审批手续。Ⅲ期临床试验为扩大临床试验,病例数一般不少于300例,在多家医院或全国范围内进行,有的在国际范围内进行。目的是在较大范围内对新药的疗效、适应症、不良反应、药物相互作用等进行评价。Ⅳ期临床试验为上市后临床试验或称上市后药物监察,目的是对已在临床上广泛应用的新药进行社会性考察,发现推广应用后可能出现的毒副反应和发现新的治疗用途,重点是新药的不良反应监察。此外,还包括未能在上市前进行的某些特殊病人的安全有效性考察,如新药在老年人、幼儿、孕妇、肝肾功能异常等病人的临床试验应在肯定新药安全有效并已批准上市后进行(专用于老人、小儿或终止妊娠等新药除外)。以上为英、美等国家的四期分期法,我国通常分三期,其中Ⅱ期相当于国外的Ⅱ、Ⅲ期,我国新药上市后的Ⅲ期试验相当于国外的Ⅳ期试验。
2 上市药物再评价
上市药物再评价包括两类:一类是针对上市药品所存在的问题(如疗效差或毒性较大等)进行临床对比研究,也可先做实验对比研究,然后再进行临床对比验证。另一类是进行流行病学调研,对再评价品种的安全有效性进行评价。药品再评价是临床药理研究单位的经常性工作之一,许多安全有效的新品种不断问世,对某些相形见拙的有必要进行研究和再评价,为药品研制、管理及使用部门决定继续使用或减量生产或淘汰这些品种提供科学依据。比如,四环素再评价研究证实,部分分离的耐四环素菌株达90%以上,引起有关领导部门重视,即减少了四环素的产量,调整了抗生素研究与生产的品种结构。经常对市场上常用药物与新药之间进行对比研究,可发现它们之间的优缺点和作用差别,提出合理治疗方案。
3 药物不良反应(ADR)监察
据报道,药物不良反应在综合医院住院病人中的发生率为0.3~1%,监护病房为3%,因此,药物不良反应监察是临床药理研究单位的一项经常性任务,各国卫生领导部门都极其重视这项工作。由于ADR的危害具有国际性,1967年开始建立了ADR国际监察系统,进行研究工作并指导各国ADR&127监察系统,目前我国有全国和全军ADR监察系统,全军ADR监察中心设在301医院,各基地每年向中心呈报ADR监察资料,根据系统所获得的资料,经电脑分析处理,可及时发现ADR发生率高和程度严重的药物,并控制或淘汰,保证用药者的安全。比如,在英国ADR监察实行黄卡系统,全国医务人员或其他有关人员发现药物不良反应立即填写黄卡向英国医药安全委员会(The Committee on Safety of Medicines CSM)报告。镇痛药异丁苯乙酸(ibufenac),就是根据黄卡系统所获得的资料,由于其肝毒性明显而被淘汰。
相关思维导图模板

树图思维导图提供 随机森林回归工作原理 在线思维导图免费制作,点击“编辑”按钮,可对 随机森林回归工作原理 进行在线思维导图编辑,本思维导图属于思维导图模板主题,文件编号是:a98e4f3d9d374a7681e0ca8c59dc8ebf

树图思维导图提供 第一章 传播与人、社会 在线思维导图免费制作,点击“编辑”按钮,可对 第一章 传播与人、社会 进行在线思维导图编辑,本思维导图属于思维导图模板主题,文件编号是:480261aa3db5da07188cd078e2b51497







 上海工商
上海工商